FrankSense · Reproductive Genetics · Vol.110
Can PGT-M PreventMonogenic DiseaseTransmission?
The answer is not whether the father is single.It depends on inheritance pattern, egg-source genetics,and whether a reliable PGT-M test can be built.
Inheritance comes firstAD, AR, X-linked and mosaic risk models are different.
PGT-M reduces riskIt targets a known familial variant; it is not whole-genome clearance.
Confirmation still mattersCVS, amniocentesis or postnatal targeted testing should be discussed.

Bottom Line
If a man carries or is affected by a pathogenic variant for a monogenic disorder, will his future child inherit the condition through assisted reproduction? The responsible answer is not a simple yes or no.
The word carrier means different things in different disorders. In many autosomal recessive conditions it means one variant and no disease. In autosomal dominant disease it may mean the person is affected. In X-linked recessive disease, an affected male is usually hemizygous rather than a silent carrier.
This article assumes that the paternal variant has been confirmed by clinical molecular testing, the egg source has had carrier screening, fertilization uses ICSI, and PGT-M is designed only for the known familial variant.
Inheritance first
01 | Start With Inheritance: Will the Child Be Affected?
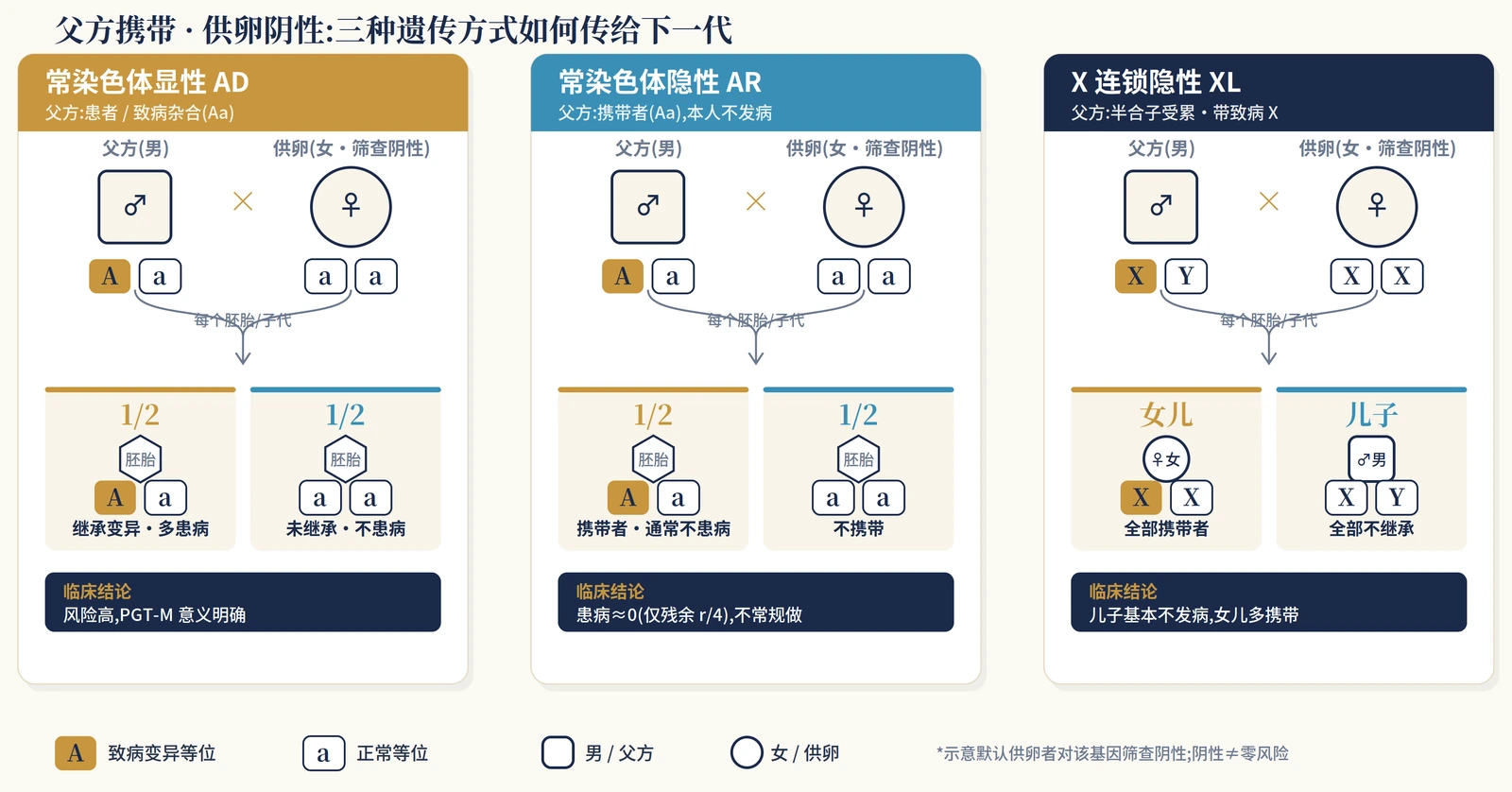
This step is more basic than deciding whether to use PGT-M. The common counseling error is to treat “inherits the variant” and “will be clinically affected” as the same statement; in X-linked disease, reduced penetrance and screened egg-source scenarios, those two statements may be very different.
Once X-linked dominant disease, de novo variants and mosaicism are added, the risk model becomes a practical decision table rather than a simple yes-or-no answer.
| Scenario | Typical paternal status | Per-pregnancy conclusion if the egg source screens negative | PGT-M relevance |
|---|---|---|---|
| Autosomal dominant | Usually heterozygous and affected | About 50% of embryos inherit the paternal variant; high-penetrance disorders may mean about 50% affected risk | High risk, clear utility |
| Autosomal recessive | Typical carrier | If the egg source truly does not carry the same gene, the child is usually not affected; about 50% may be carriers | ASRM does not recommend routine PGT-M when only one asymptomatic partner is a carrier |
| X-linked recessive | Hemizygous affected male, not a silent carrier | Daughters inherit the paternal X and are often carriers; sons inherit the Y and do not inherit that paternal X variant | Inheriting the variant is not the same as having typical disease |
| X-linked dominant | Affected or carrying a pathogenic X | Daughters inherit the paternal X and are usually high risk; sons do not inherit the paternal X | Daughter risk is high |
| Apparently de novo in the father | Depends on the genetic mechanism | If the variant is present in the germline, transmission follows the relevant AD or X-linked model | The key question is whether sperm carry the variant |
| Paternal / gonadal mosaicism | Only some sperm carry the variant | Risk is no longer the classic 50%; it depends on the proportion of variant-carrying sperm | Individual modelling is required |
Two points are especially easy to miss. First, a de novo variant that arose in the father does not automatically make offspring risk low. If the variant is present in the germline, transmission can still be substantial. Second, paternal gonadal mosaicism should not be forced into a textbook 50% model; the risk depends on how much of the sperm population carries the variant.
PGT-M workflow
02 | What PGT-M Does: Reduce a Known Familial Risk
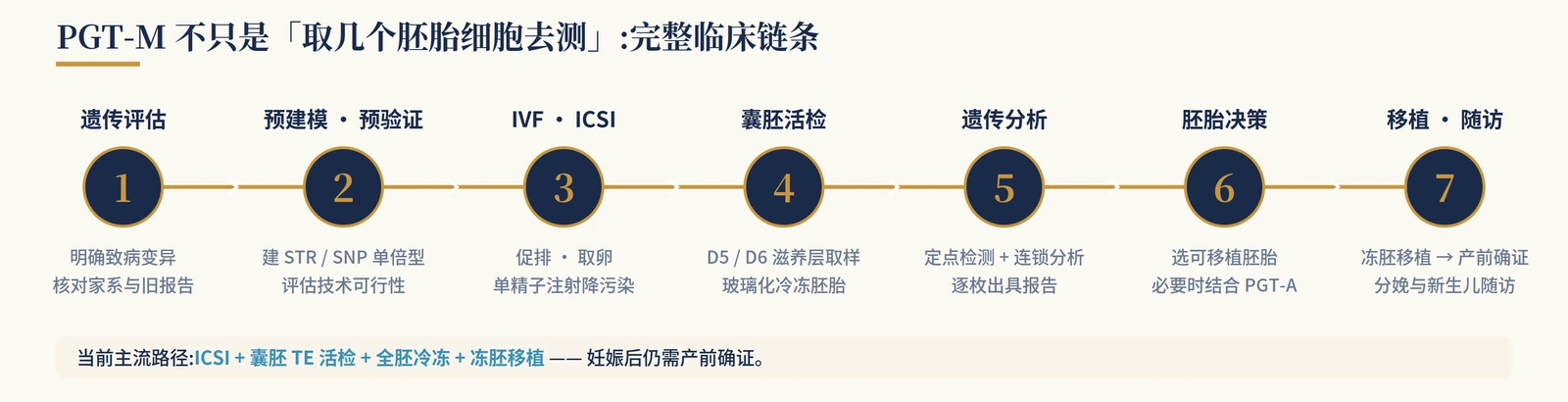
PGT-M is meant to reduce transmission of a known monogenic condition when the familial pathogenic variant is clear and a reliable assay can be built.
It is not whole-genome clearance and should not be marketed as a universal way to improve pregnancy rates. HFEA public information lists more than 2,000 approved genetic conditions for PGT-M in the UK, while ASRM notes that many Mendelian disorders with a known genetic cause may be considered, subject to feasibility, family samples and haplotype construction.
The clinical chain usually runs from genetic counseling and record review to family sample collection, haplotyping or linkage analysis, IVF/ICSI, blastocyst biopsy, genetic analysis, embryo decision-making, prenatal confirmation and newborn follow-up.
| Method / combination | Principle | Typical use | Accuracy and limits |
|---|---|---|---|
| Direct mutation testing | Tests the known pathogenic site | Clear and technically simple variant | When used alone, more vulnerable to ADO and contamination; rarely ideal as the only evidence |
| Direct testing + STR/SNP linkage | Tests the variant and surrounding haplotype | One of the most common robust routes | Reduces single-site misclassification; needs informative markers and often family samples |
| Karyomapping / SNP haplotyping | Genome-wide SNP-based haplotype construction | Family samples are available and a broader haplotype approach is useful | More robust against ADO and contamination; recombination still needs careful interpretation |
| WGA + targeted NGS / combined approaches | Amplify first, then perform targeted analysis | Comprehensive reporting after trophectoderm biopsy | Widely used; cannot eliminate mosaicism, recombination or sample identity errors |
| PGT-M + PGT-A | Adds aneuploidy information to PGT-M | Older egg source, repeated failures, clinic-specific indications | More information, but not routine for everyone; may reduce the number of transferable embryos |
Reliability comes from variant confirmation, family modelling, linkage checks and laboratory quality control, not from a single site being tested in isolation.
Accuracy and residual risk
03 | High Accuracy Is Not Zero Risk
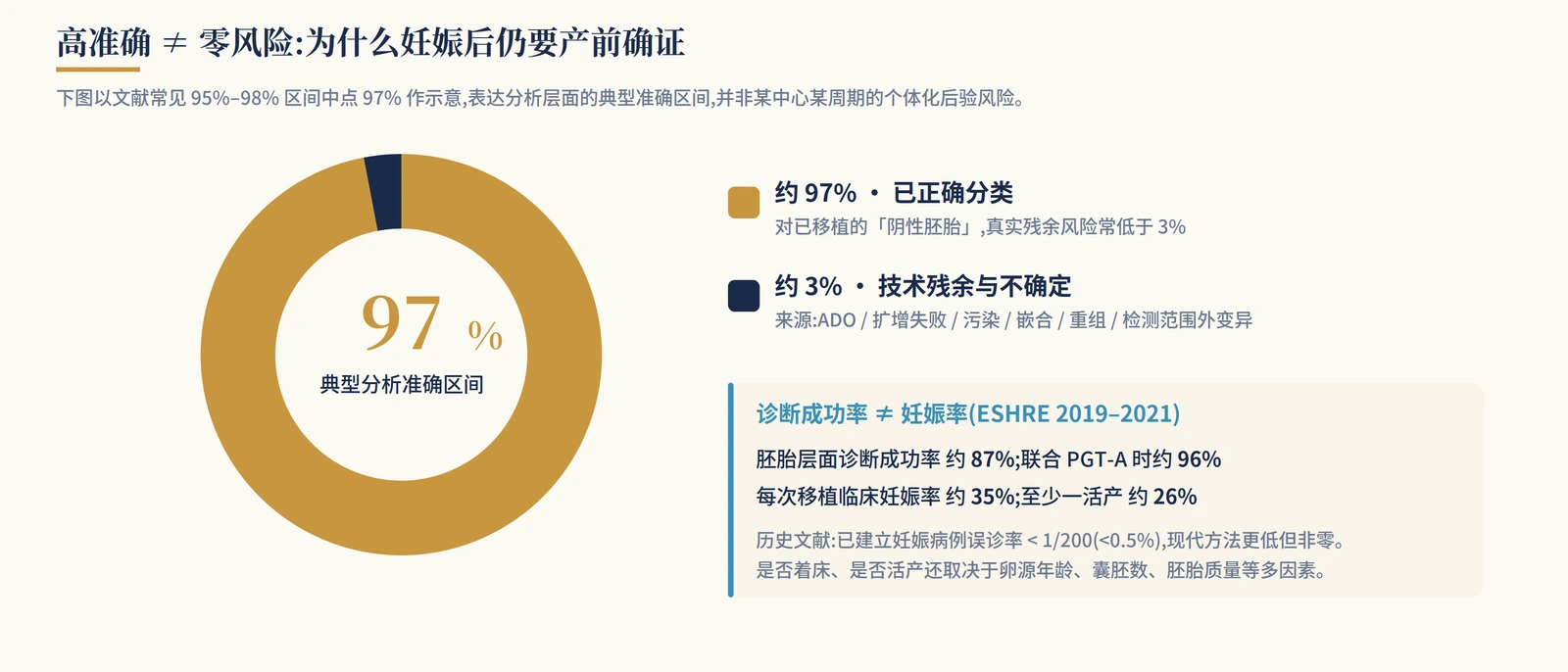
ACOG and ASRM both emphasize that false-positive and false-negative results can occur in PGT. A negative or unaffected PGT-M result does not rule out every genetic problem in a newborn, and it does not cover microdeletions, microduplications, de novo variants, imprinting disorders or conditions outside the designed assay.
How accurate is it? Published summaries and center-level reports vary, but a common broad range is about 95%-98%. For established pregnancies, historical misdiagnosis has been reported below 1/200 (<0.5%), and modern approaches may be lower. That is still not zero and does not remove the need for prenatal confirmation.
Residual risk can come from allele drop-out, amplification failure, contamination, embryo or placental mosaicism, meiotic or mitotic recombination, sample identity error, incorrect donor or family information, and variants outside the tested region. Diagnostic success is also not the same as pregnancy rate or live-birth rate; implantation and live birth still depend on egg-source age, blastocyst number and embryo quality.
Should PGT-A be added?
The argument for adding PGT-A is stronger when the egg source is older, prior cycles produced few usable embryos, or the goal is to avoid transferring embryos that are monogenic-variant-negative but clearly aneuploid. The cautious view is that ACOG and ASRM do not support PGT-A as a routine add-on for every IVF patient. In donor-egg settings, benefit must be judged by embryo number, clinic policy and patient goals. Mosaic embryo reporting and a smaller transferable embryo pool also need to be discussed before testing.
Probability
04 | Probability: Embryo Number Changes the Chance
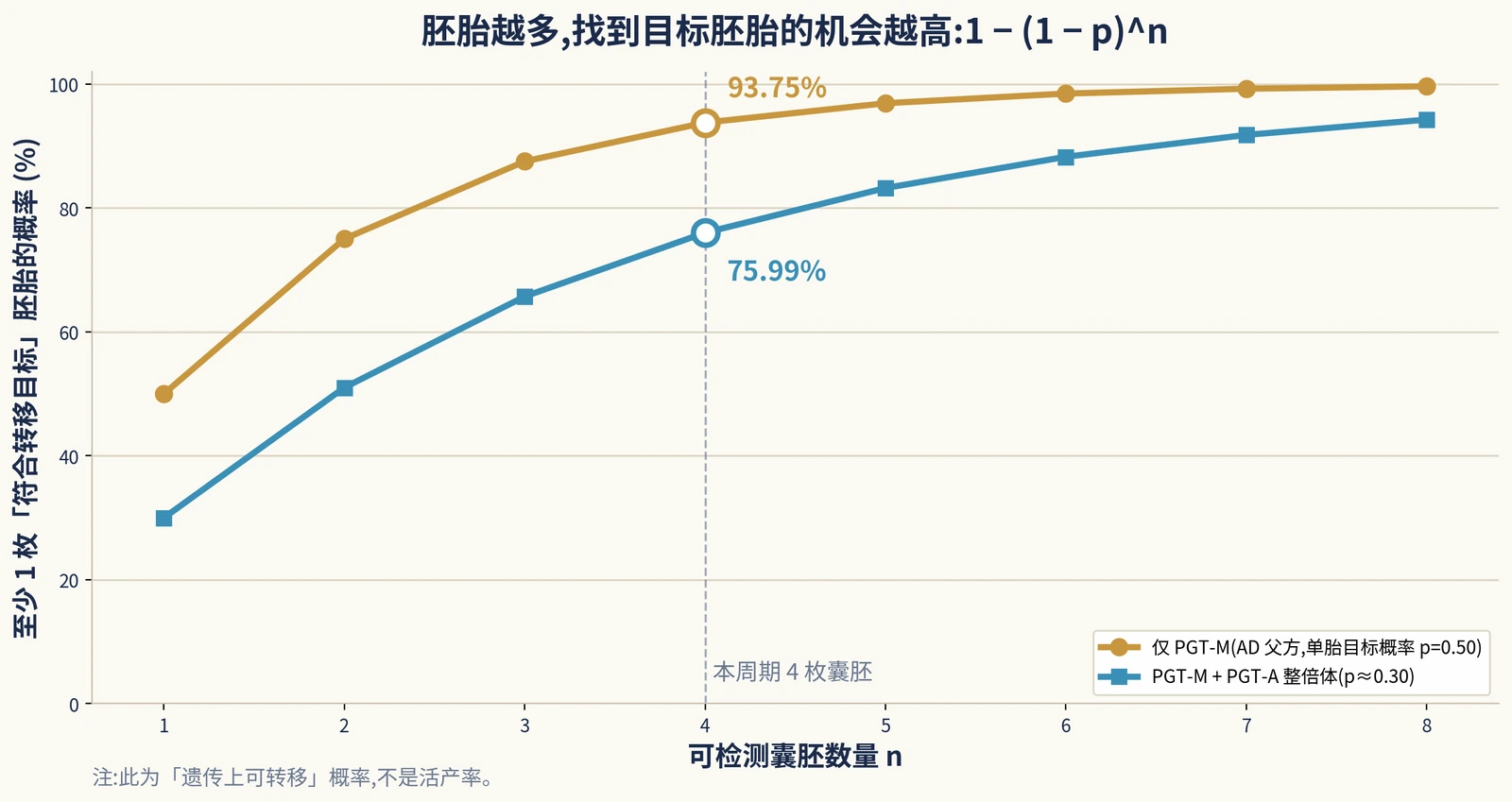
The practical formula is simple: each embryo has a probability p of meeting the transfer target; among n embryos, the probability of at least one target embryo is 1 - (1 - p)n.
Here, p may mean “not carrying the paternal AD variant,” or it may mean “both variant-free and euploid.” If euploidy is included, the extra assumption must be stated clearly. This is basic probability, not a live-birth promise.
Case 1: a 35-year-old autosomal dominant carrier
The core issue is not that he is single; it is that the father is heterozygous for an AD disorder, such as NF1 or another high-penetrance condition, and plans IVF/ICSI with a screened egg source and PGT-M.
Each embryo has about a 1/2 chance of inheriting the paternal pathogenic variant and a 1/2 chance of not inheriting it. With four testable blastocysts, the chance of at least one variant-free embryo is 1 - 0.54 = 93.75%. If, as a teaching assumption, euploid probability is 60% and independent of the monogenic locus, the chance of an embryo being both variant-free and euploid is about 0.30; four embryos give 1 - 0.74 = 75.99%.
Counseling language: your baseline genetic risk is not vague; each embryo has roughly a 50% chance of inheriting the paternal variant. PGT-M can markedly reduce that classic risk, but it is not a 100% guarantee. Even after transferring an embryo reported as unaffected, prenatal confirmation is still recommended because rare misdiagnosis, mosaicism and untested variants remain possible.
Case 2: hemophilia A, an X-linked recessive condition
This case shows why “inherits the variant” and “has typical disease” are different. An affected father produces sperm carrying either X or Y. Daughters receive the paternal X; sons receive the paternal Y. If the egg source does not carry the relevant F8 variant, daughters usually carry the paternal variant, while sons usually do not inherit that paternal X-linked variant.
With a roughly even sex ratio, about half of children may carry the variant. But if the question is whether a child will have typical male hemophilia, the answer is different: sons are usually not affected by that paternal X variant, while daughters are usually carriers. Female carriers may still have clinical relevance, including bleeding symptoms in some cases because of skewed X inactivation.
Counseling language: in an X-linked recessive condition, daughters receive your X and sons do not. If the egg source does not carry the same gene variant, sons are usually unaffected, while daughters usually become carriers. The family must decide whether the goal is to avoid typical disease or also to avoid carrier status; those goals lead to different embryo-selection strategies.
Two additional common situations
Why single-partner AR carrier status often is not worth PGT-M: if the egg source has a residual carrier risk r after expanded screening, affected-child risk is approximately paternal transmission 1/2 × residual egg-source risk r × maternal transmission 1/2 = r/4. If r = 1/1000, affected risk is about 1/4000, or 0.025%.
How to model paternal gonadal mosaicism: if about 8% of sperm carry a particular AD variant, the chance of transmission is closer to 8% than to the classic 50%. The right wording is not simply “low risk,” but “not the classic 50%, yet still above background risk and requiring individual modelling.”
Ethics, law and cost
05 | Ethics, Law and Cost: Feasible Does Not Always Mean Appropriate

The strongest ethical indication for PGT-M remains serious, clear and verifiable monogenic disease. Its use has expanded into adult-onset disease, reduced penetrance and HLA matching, where the question shifts from “can it be done” to “should it be done.” Non-directive genetic counseling, patient autonomy and parallel discussion of alternatives are central.
Whether to transfer an embryo classified as affected is not answered by a slogan. It touches reproductive autonomy, the welfare of the future child, professional conscience and family context, and it requires clinic policy, informed consent and case-by-case judgment.
Regulation varies. The United States has no single national PGT-M regulator, while the UK HFEA requires approved conditions and licensed clinics. Sex information is generally limited to medical indications in many jurisdictions.
| Cost item | Typical range | Comment |
|---|---|---|
| IVF / ICSI cycle | US about USD 15,000-25,000+; UK private about GBP 4,890-5,520 | Medication, region and lab package vary |
| PGT-M lab fee | US about USD 7,000-12,000 / cycle; UK private often from GBP 3,875 | Usually higher than PGT-A because custom test development is needed |
| Embryo biopsy / added analysis | Hundreds to thousands of dollars or pounds | Bundling and per-embryo pricing vary |
| PGT-A add-on | About USD 2,000-5,000+ | Should not be assumed for everyone |
| Frozen embryo transfer | UK about GBP 1,800-2,000; often separate in the US | Many PGT-M cases require freezing while results are pending |
| Egg donation | US about USD 10,000-30,000+ for the egg-source component alone | Often a major driver of total cost |
Timing is also longer than a standard IVF cycle. Genetic counseling and test development come first, followed by stimulation, biopsy, results and frozen embryo transfer. First-round commercial test development may take about 3-16 weeks; later cycles can be faster once the model is built.
Pregnancy and follow-up
06 | Pregnancy and Newborn Follow-Up

Once pregnancy is established, prenatal diagnosis should still be offered. ASRM, ESHRE and ACOG are aligned on this point: pregnancy after PGT-M should include counseling about rare misdiagnosis and the option of CVS or amniocentesis. If PGT-A was not performed, routine fetal aneuploidy screening or diagnosis should still be offered.
Follow-up should not stop at birth. If prenatal confirmation was not performed, targeted molecular testing after birth may be valuable. For X-linked disease, reduced penetrance, late-onset disease or clinically meaningful carrier status, family management and pediatric or specialty follow-up should be planned.
Current follow-up data are broadly reassuring, but should not be overstated. Available studies have not shown a major congenital malformation rate higher than IVF/ICSI controls, yet long-term outcomes by disease and testing pathway still require continued follow-up.
Responsible care links counseling, test development, embryo decisions, prenatal confirmation and newborn follow-up; it does not end on transfer day.
Conclusion
07 | One-Sentence Conclusion
Whether a man who carries or is affected by a monogenic variant will pass disease to a child depends on inheritance pattern, whether he is truly affected or only a carrier, egg-source genetics and whether a suitable PGT-M assay can be built.
The most common mistake is treating “only one partner is an AR carrier” and “the father is affected by AD or X-linked disease” as the same problem. The former often carries low affected-child risk after egg-source screening; the latter may carry a real vertical transmission risk and stronger PGT-M utility.
PGT-M can substantially reduce transmission of a known familial variant, but it is not a 100% block against all genetic, chromosomal, pregnancy or newborn risks.
FAQ
Will a child definitely inherit a paternal monogenic variant?
No. The answer depends on inheritance pattern, egg-source genotype and whether the father is affected, a carrier or mosaic.
Can PGT-M guarantee a child without genetic disease?
No. It targets a known familial variant and still has residual risk.
Is PGT-M needed when only the father is an autosomal recessive carrier?
Usually not if the egg source screens negative for the same gene and the father is asymptomatic; ASRM lists this as a non-recommended indication.
Is prenatal diagnosis still needed after PGT-M?
Patients should be offered prenatal confirmation by CVS or amniocentesis where appropriate.
Guidelines and Sources
This page summarizes public guidelines, regulatory information and professional recommendations for patient education and counseling.
- 胚胎植入前遗传学检测的遗传咨询专家共识. 中华医学会围产医学分会. 2025.
- 单基因病胚胎着床前遗传学检测专家共识. 中国医师协会生殖医学专业委员会 / 医学遗传医师分会. 2021.
- 胚胎植入前遗传学诊断 / 筛查技术专家共识. 2018.
- 血友病的胚胎着床前遗传学检测专家共识. 2021.
- ASRM Practice Committee: Indications and management of PGT-M, 2023 Source
- ESHRE PGT Consortium good practice recommendations for PGT-M, 2020 Source
- HFEA: Approved PGT-M and PTT conditions Source
- ACOG Committee Opinion No. 799: Preimplantation Genetic Testing Source
- ACOG: Carrier Screening in the Age of Genomic Medicine Source
- Hardy K, et al. Role of prenatal diagnosis after PGT; historical PGT-M misdiagnosis rate < 1/200. 2020.
- Poulton A, et al. Review: PGT-M accuracy approximately 95%-98%. 2025.
- Spinella F, et al. ESHRE PGT Consortium data 2019-2021.
- De Rycke M. PGT-M status review: technical evolution and offspring follow-up. 2020.
Turn a genetic report into a practical pathway
Bring the variant, inheritance pattern, family samples, egg-source screening and embryo count into one PGT-M feasibility review.
Review the pathwayThis article is educational and does not replace personal medical care, genetic counseling or laboratory-specific test review.